Viral Sequence Feature Extraction Tool
Welcome to ViSFE!
ViSFE - A tool for feature extraction of viral genome sequences in NGS data. ViSFE overcomes the technical challenge of random fragmented reads and is a universal algorithm for analyzing viral genome NGS data. The website provides online tutorials as well as code for localized operation (current version :2024-8-10).
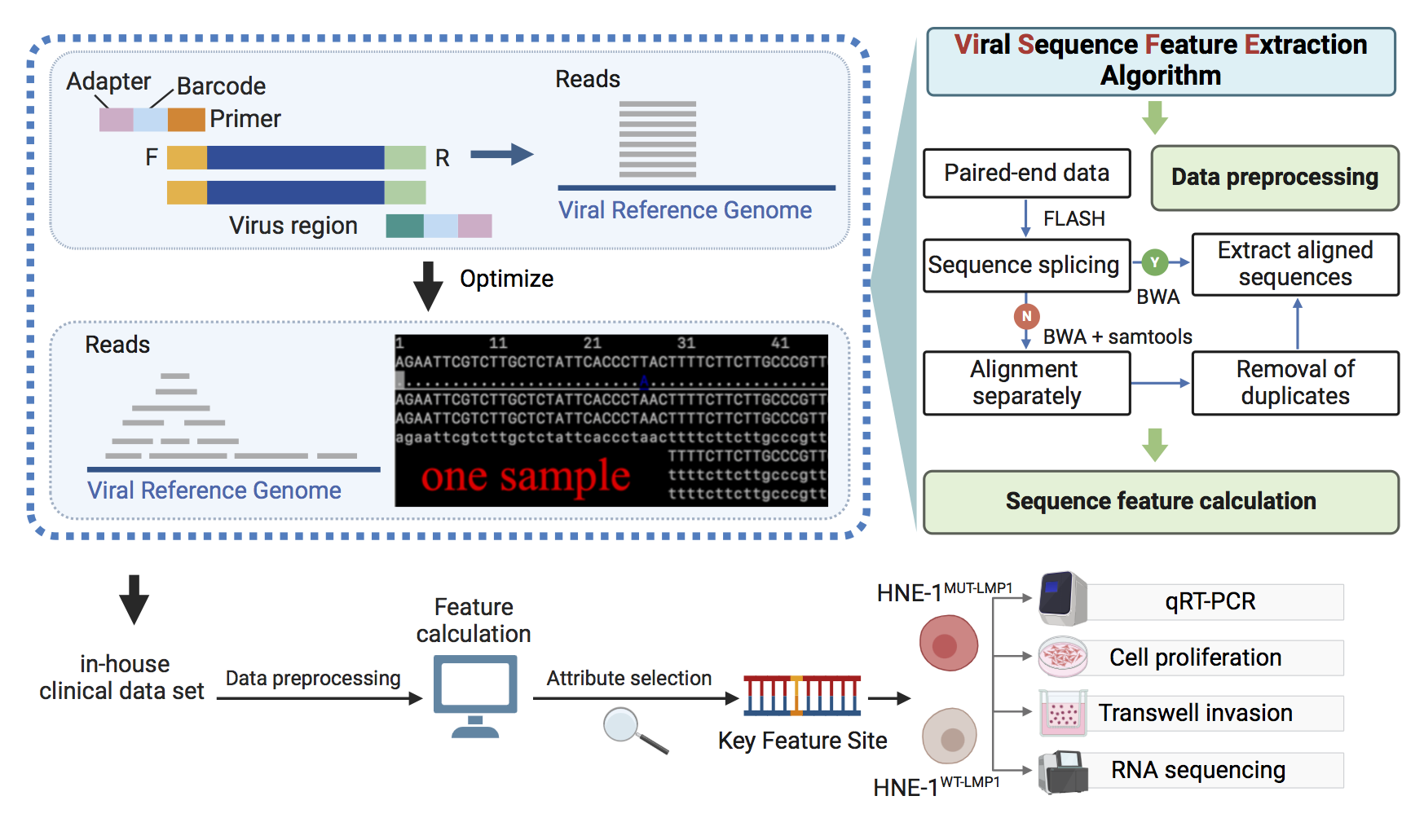
What is ViSFE algorithm?
ViSFE can generate mutation features and Shannon entropy features of viral sequences at the nucleotide level from the raw fastq sequence data obtained by NGS, through steps such as sequence cleaning, alignment, assembly, and feature calculation. ViSFE addresses the issue of extracting viral sequence features from amplicon-based NGS data, and can generate a one-stop sequence site feature matrix.
How do the researchers/
clinicians can benefit from
ViSFE?
By leveraging publicly available data resources, it can help researchers/clinicians quickly obtain new clues for further experimental research and clinical validation. ViSFE can process viral genomic data from different laboratories with various formats, solving issues such as uneven sequence lengths and data format discrepancies. It can effectively handle public, fragmented sequence data, facilitating subsequent model establishment and prediction, thereby maximizing the value of such data, reducing the time and manpower required for acquiring and analyzing high-quality data, and promoting a faster process of viral research.
Our previous work and resources
Browse other work, database and tools
1.Quasispecies pattern of hepatitis B virus predicts hepatocellular carcinoma
2.HBV quasispecies features in HBsAg+/HBsAb+ patients with HBV genotype C
3.Unique Mutation Pattern of HPV16 and Predicts Cervical Cancer
4.ViMIC: a database of human disease-related virus mutations, integration sites and cis-effects
5.ViMRT: a text-mining tool and search engine for automated virus mutation recognition